The Glucosylceramidase beta (GBA) Monoclonal Antibody (CAB19057) is a high-quality antibody developed for reliable detection and analysis of target proteins. This antibody, developed through rabbit immunization, exhibits high specificity and sensitivity for detecting GBA protein in various samples, particularly in human tissues. Validated for use in applications such as Western blot, immunofluorescence, and immunohistochemistry, this antibody allows for precise and reliable detection of GBA expression in cell lines and tissues.GBA, also known as glucocerebrosidase, is essential for the breakdown of glucosylceramide, a lipid molecule implicated in several lysosomal storage disorders, including Gaucher disease.
This antibody is validated for use in WB, IHC-P, ELISA applications and has demonstrated reactivity against Human, Rat samples.
Product Name:
Glucosylceramidase beta (GBA) Monoclonal Antibody
SKU:
CAB19057
Size:
20μL, 100μL
Reactivity:
Human, Rat
Clone Number:
ARC0500
Conjugate:
Unconjugated
Immunogen:
Synthetic peptide. This information is considered to be commercially sensitive.
This gene encodes a lysosomal membrane protein that cleaves the beta-glucosidic linkage of glycosylceramide, an intermediate in glycolipid metabolism. Mutations in this gene cause Gaucher disease, a lysosomal storage disease characterized by an accumulation of glucocerebrosides. A related pseudogene is approximately 12 kb downstream of this gene on chromosome 1. Alternative splicing results in multiple transcript variants.
Purification Method
Affinity purification
Gene ID
2629
RRID
AB_2862550
Buffer Information
Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS containing 50% glycerol and 0.05% BSA, preserved with proclin300 or sodium azide, pH 7.3.
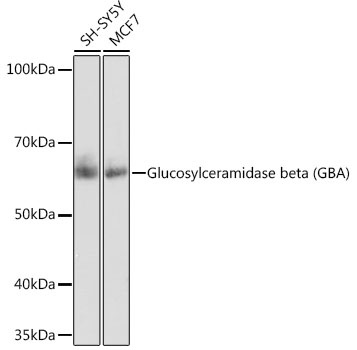
Western blot analysis of various lysates using Glucosylceramidase beta (GBA) Rabbit mAb (CAB19057) at 1:1000 dilution. Secondary antibody: HRP-conjugated Goat anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25μg per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (AbGn00020). Exposure time: 10s.
Western blot analysis of lysates from Rat brain, using Glucosylceramidase beta (GBA) Rabbit mAb (CAB19057) at 1:1000 dilution. Secondary antibody: HRP-conjugated Goat anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25μg per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (AbGn00020). Exposure time: 3min.
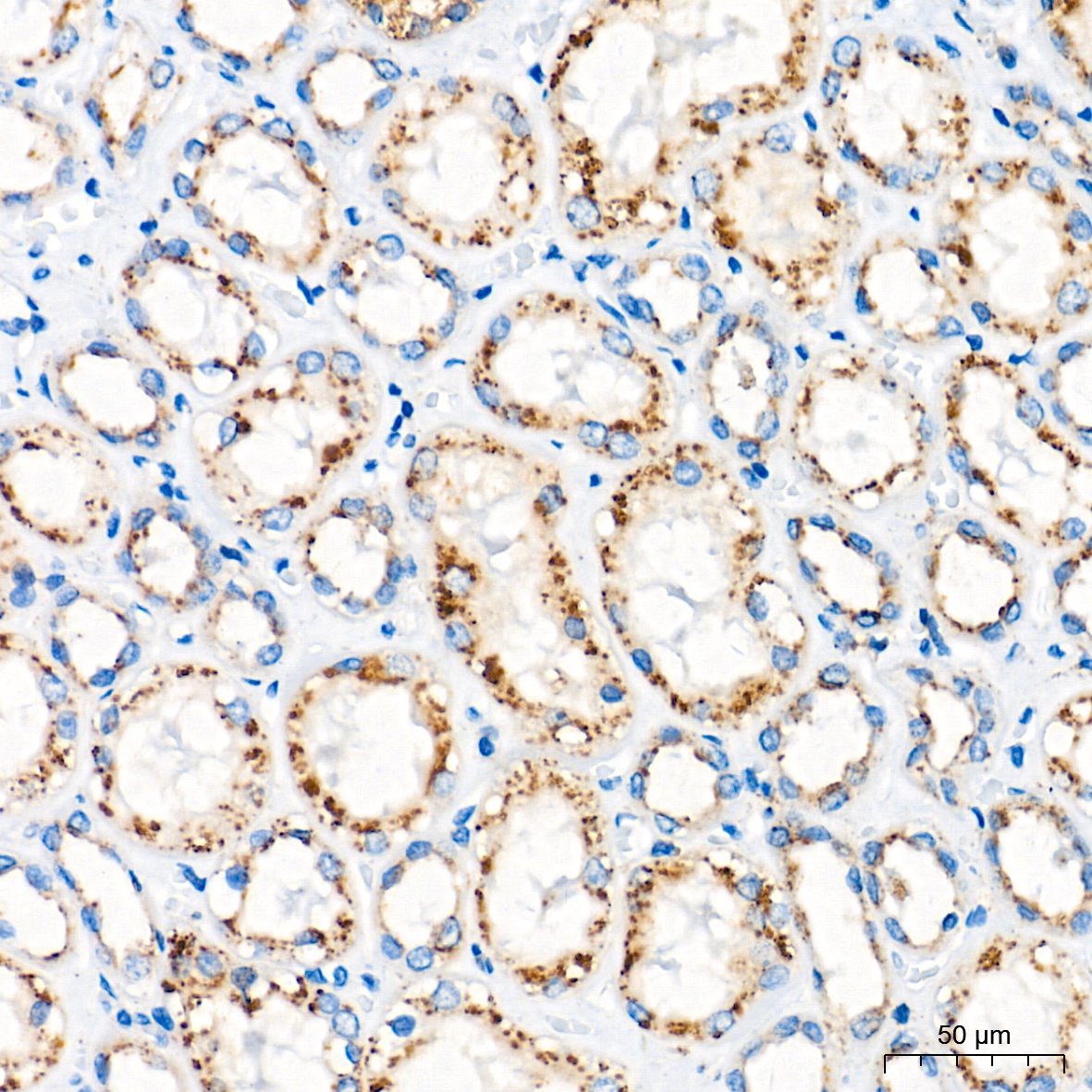
Immunohistochemistry analysis of paraffin-embedded Human kidney tissue using Glucosylceramidase beta (GBA) Rabbit mAb (CAB19057) at a dilution of 1:400 (40x lens). High pressure antigen retrieval performed with 0.01M Tris-EDTA Buffer (pH 9.0) prior to IHC staining.
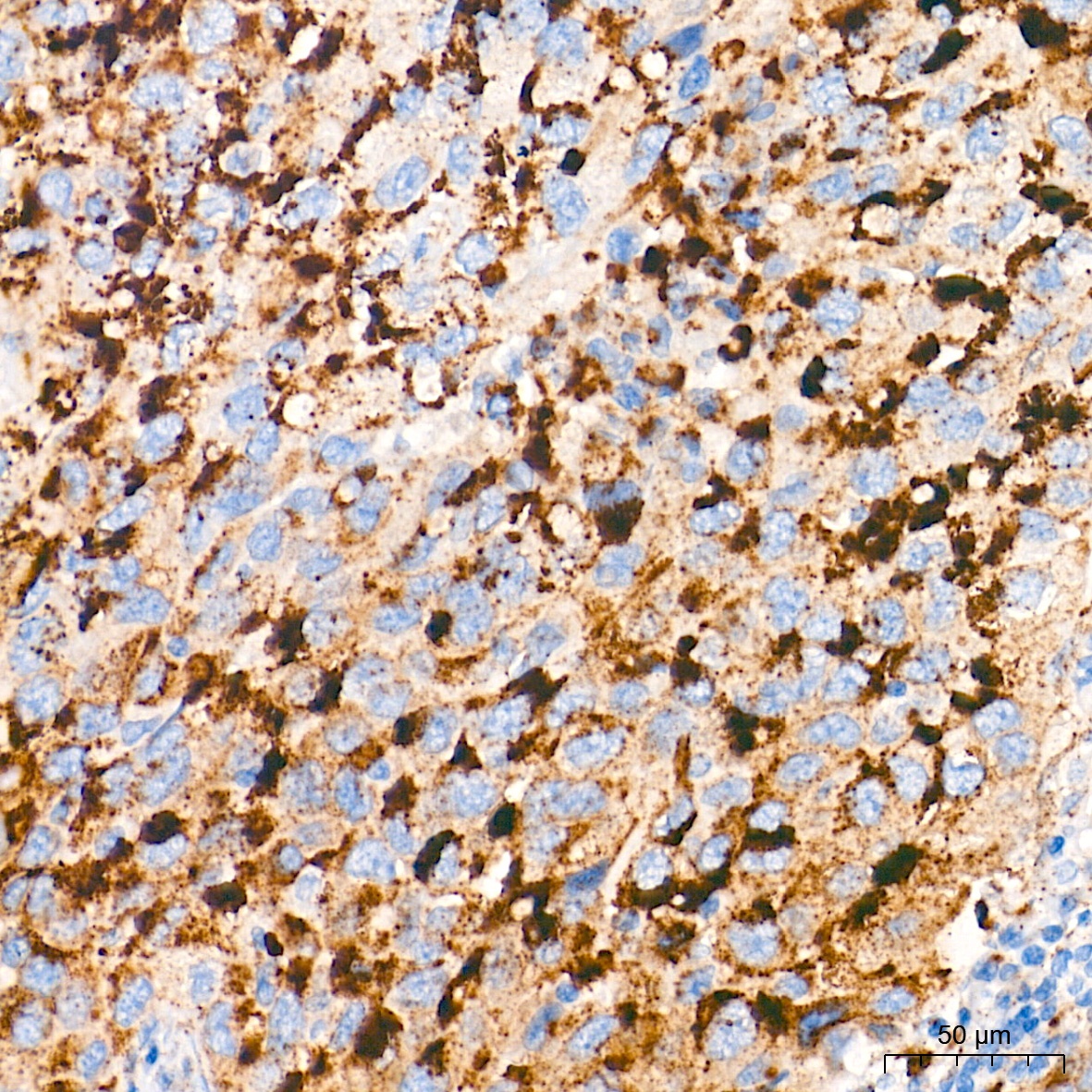
Immunohistochemistry analysis of paraffin-embedded Human lung squamous carcinoma tissue using Glucosylceramidase beta (GBA) Rabbit mAb (CAB19057) at a dilution of 1:400 (40x lens). High pressure antigen retrieval performed with 0.01M Tris-EDTA Buffer (pH 9.0) prior to IHC staining.
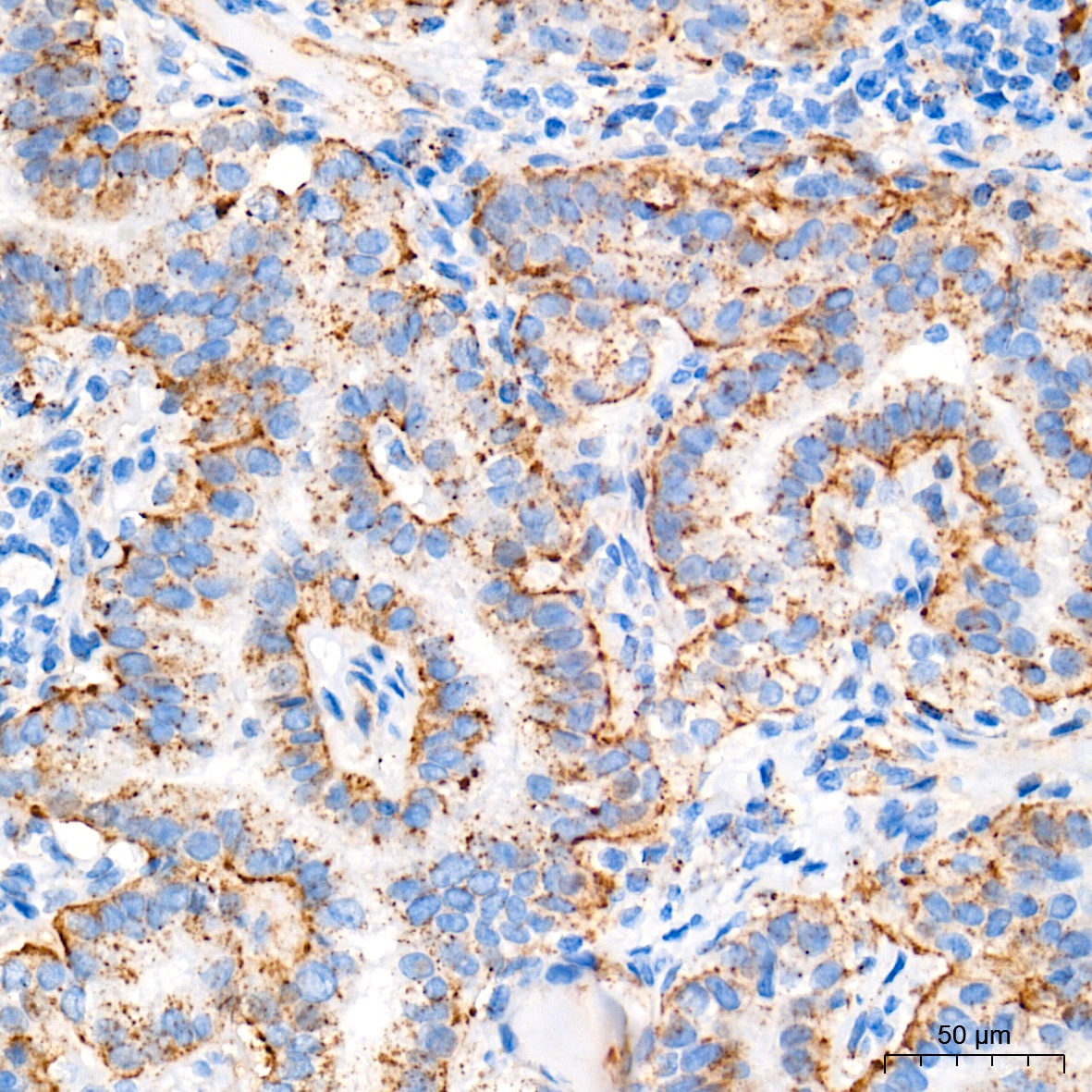
Immunohistochemistry analysis of paraffin-embedded Human thyroid cancer tissue using Glucosylceramidase beta (GBA) Rabbit mAb (CAB19057) at a dilution of 1:400 (40x lens). High pressure antigen retrieval performed with 0.01M Tris-EDTA Buffer (pH 9.0) prior to IHC staining.
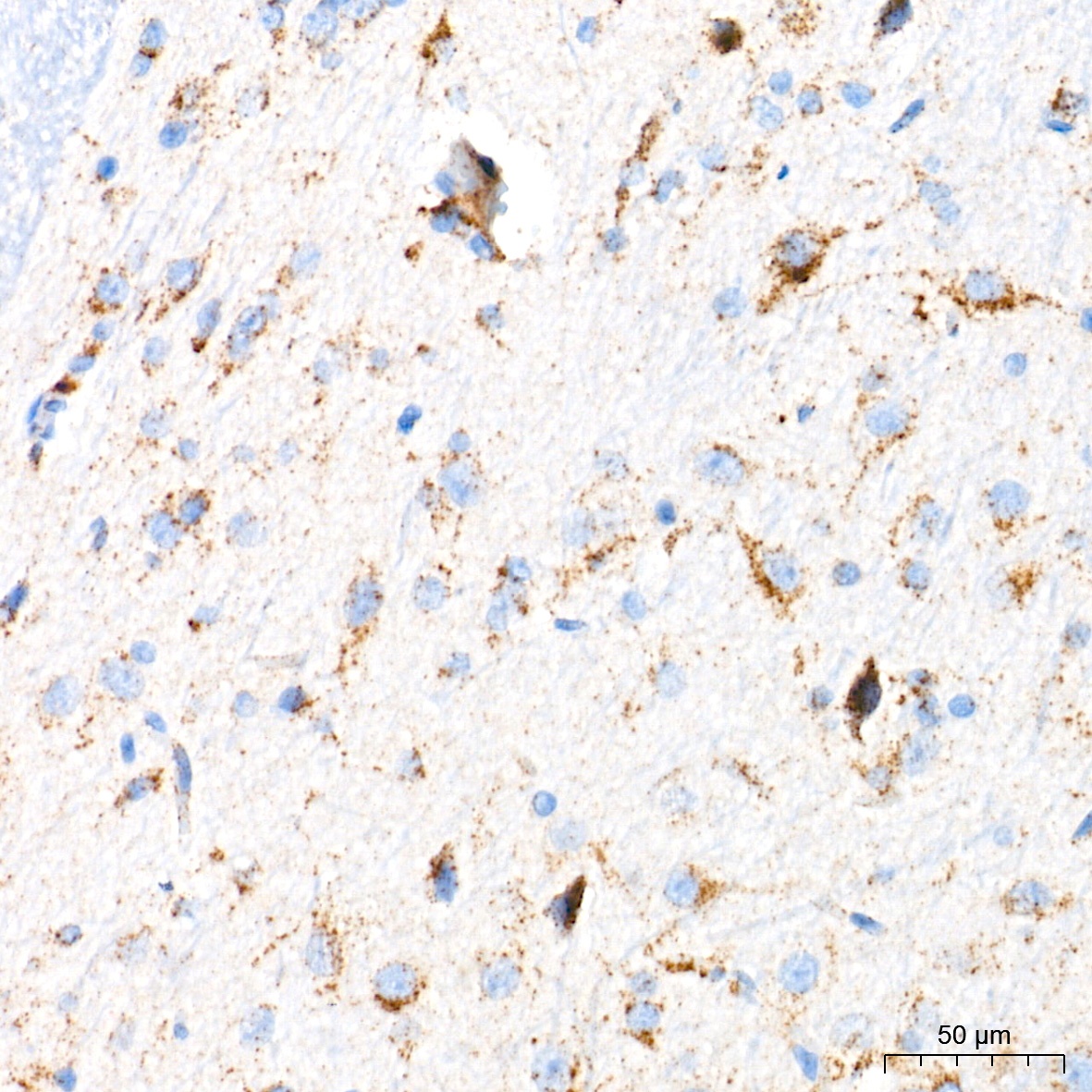
Immunohistochemistry analysis of paraffin-embedded Rat brain tissue using Glucosylceramidase beta (GBA) Rabbit mAb (CAB19057) at a dilution of 1:400 (40x lens). High pressure antigen retrieval performed with 0.01M Tris-EDTA Buffer (pH 9.0) prior to IHC staining.
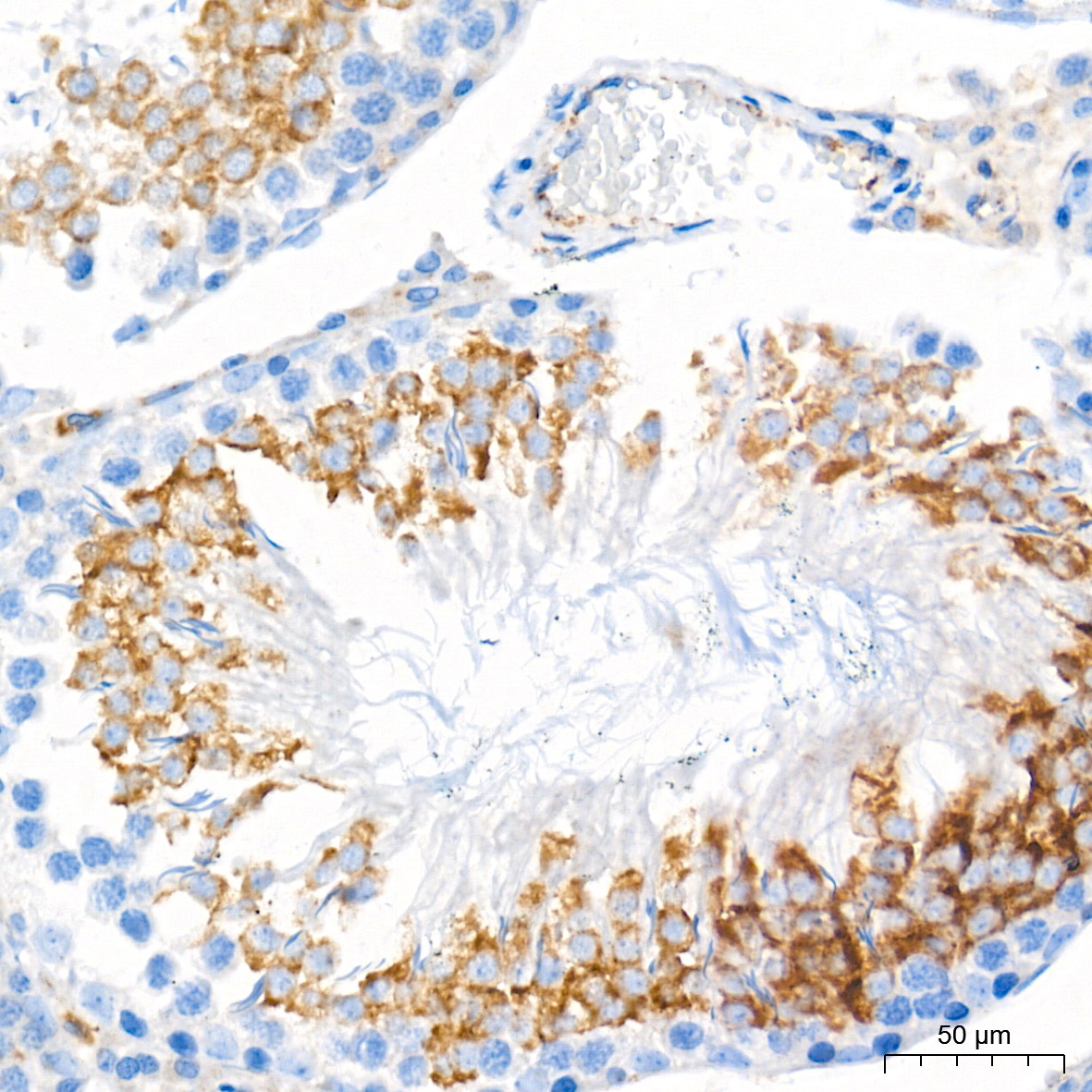
Immunohistochemistry analysis of paraffin-embedded Rat testis tissue using Glucosylceramidase beta (GBA) Rabbit mAb (CAB19057) at a dilution of 1:400 (40x lens). High pressure antigen retrieval performed with 0.01M Tris-EDTA Buffer (pH 9.0) prior to IHC staining.