The RP2 Polyclonal Antibody (PAC04413) is a valuable tool for researchers studying RP2, a protein involved in ciliary function and linked to retinitis pigmentosa. This antibody, produced in rabbits, demonstrates high specificity for human samples and has been validated for use in Western blot applications. By binding to the RP2 protein, it allows for accurate detection and analysis in a variety of cell types, making it an essential component for studies in ophthalmology and genetic disorders.RP2 is a crucial player in cilia biology, playing a role in ciliary trafficking and maintenance.
Mutations in the RP2 gene are associated with retinitis pigmentosa, a progressive vision disorder that leads to blindness. Research into the function and regulation of RP2 is essential for understanding the mechanisms underlying this disease and developing potential treatment options. The RP2 Polyclonal Antibody provides researchers with a tool to investigate the role of RP2 in cellular processes and disease pathology, furthering our knowledge in the field of genetic eye disorders.
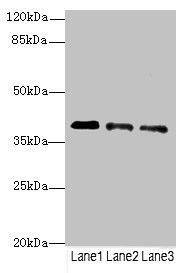
Western blot. All lanes: RP2antibody at 3.81µg/ml. Lane 1: Jurkat whole cell lysate. Lane 2: HepG2 whole cell lysate. Lane 3: Hela whole cell lysate. Secondary. Goat polyclonal to rabbit IgG at 1/10000 dilution. Predicted band size: 40 kDa. Observed band size: 40 kDa..
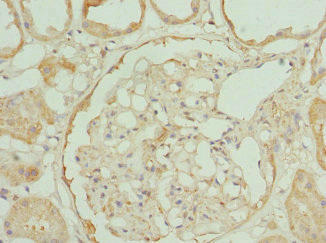
Immunohistochemistry of paraffin-embedded human kidney tissue using PACO44413 at dilution of 1:100.
Background:
Acts as a GTPase-activating protein (GAP) involved in trafficking between the Golgi and the ciliary membrane. Involved in localization of proteins, such as NPHP3, to the cilium membrane by inducing hydrolysis of GTP ARL3, leading to the release of UNC119 (or UNC119B). Acts as a GTPase-activating protein (GAP) for tubulin in concert with tubulin-specific chaperone C, but does not enhance tubulin heterodimerization. Acts as guanine nucleotide dissociation inhibitor towards ADP-ribosylation factor-like proteins.
Synonyms:
Protein XRP2, RP2
UniProt Protein Function:
RP2: Acts as a GTPase-activating protein (GAP) involved in trafficking between the Golgi and the ciliary membrane. Involved in localization of proteins, such as NPHP3, to the cilium membrane by inducing hydrolysis of GTP ARL3, leading to the release of UNC119 (or UNC119B). Acts as a GTPase-activating protein (GAP) for tubulin in concert with tubulin-specific chaperone C, but does not enhance tubulin heterodimerization. Acts as guanine nucleotide dissociation inhibitor towards ADP-ribosylation factor-like proteins. Defects in RP2 are the cause of retinitis pigmentosa type 2 (RP2); also known as X-linked retinitis pigmentosa 2 (XLRP-2). RP leads to degeneration of retinal photoreceptor cells. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. Belongs to the TBCC family.Protein type: ChaperoneChromosomal Location of Human Ortholog: Xp11.3Cellular Component: centriole; Golgi apparatus; cytoplasm; plasma membrane; cytoplasmic vesicleMolecular Function: protein binding; GTP binding; unfolded protein binding; nucleoside diphosphate kinase activity; actin binding; GTPase activator activity; ATP bindingBiological Process: GTP biosynthetic process; CTP biosynthetic process; protein transport; protein folding; visual perception; organelle organization and biogenesis; UTP biosynthetic process; cell morphogenesis; nucleoside diphosphate phosphorylation; cytoskeleton organization and biogenesis; post-Golgi vesicle-mediated transport; post-chaperonin tubulin folding pathway; positive regulation of GTPase activityDisease: Retinitis Pigmentosa 2
UniProt Protein Details:
NCBI Summary:
The RP2 locus has been implicated as one cause of X-linked retinitis pigmentosa. The predicted gene product shows homology with human cofactor C, a protein involved in the ultimate step of beta-tubulin folding. Progressive retinal degeneration may therefore be due to the accumulation of incorrectly-folded photoreceptor or neuron-specific tubulin isoforms followed by progressive cell death [provided by RefSeq, Jul 2008]





")



