The CFTR Antibody (CAB8386) is a high-quality antibody developed for reliable detection and analysis of target proteins. This antibody, produced in rabbits, shows high reactivity with CFTR in human samples and is validated for use in a variety of applications, including Western blot and immunofluorescence.CFTR is a key player in maintaining the balance of ions and water across cell membranes, particularly in the respiratory and digestive systems. Mutations in the CFTR gene lead to the development of Cystic Fibrosis, a genetic disorder characterized by thick mucus production and impaired organ function.
This antibody is validated for use in WB, IF/ICC, ELISA applications and has demonstrated reactivity against Human, Mouse, Rat samples.
Product Name:
CFTR Antibody
SKU:
CAB8386
Size:
20μL, 100μL
Reactivity:
Human, Mouse, Rat
Conjugate:
Unconjugated
Immunogen:
Recombinant protein (or fragment).This information is considered to be commercially sensitive.
Cell Membrane, Early Endosome Membrane, Multi-Pass Membrane Protein.
Calculated MW:
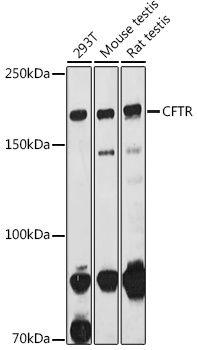
168kDa
Observed MW:
168kDa
This gene encodes a member of the ATP-binding cassette (ABC) transporter superfamily. The encoded protein functions as a chloride channel, making it unique among members of this protein family, and controls ion and water secretion and absorption in epithelial tissues. Channel activation is mediated by cycles of regulatory domain phosphorylation, ATP-binding by the nucleotide-binding domains, and ATP hydrolysis. Mutations in this gene cause cystic fibrosis, the most common lethal genetic disorder in populations of Northern European descent. The most frequently occurring mutation in cystic fibrosis, DeltaF508, results in impaired folding and trafficking of the encoded protein. Multiple pseudogenes have been identified in the human genome.
Purification Method
Affinity purification
Gene ID
1080
RRID
AB_2768875
Buffer Information
Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.09% sodium azide,50% glycerol,pH7.3.
Western blot analysis of various lysates using CFTR Rabbit pAb (CAB8386) at 1:1000 dilution. Secondary antibody: HRP-conjugated Goat anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25μg per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (AbGn00020). Exposure time: 180s.
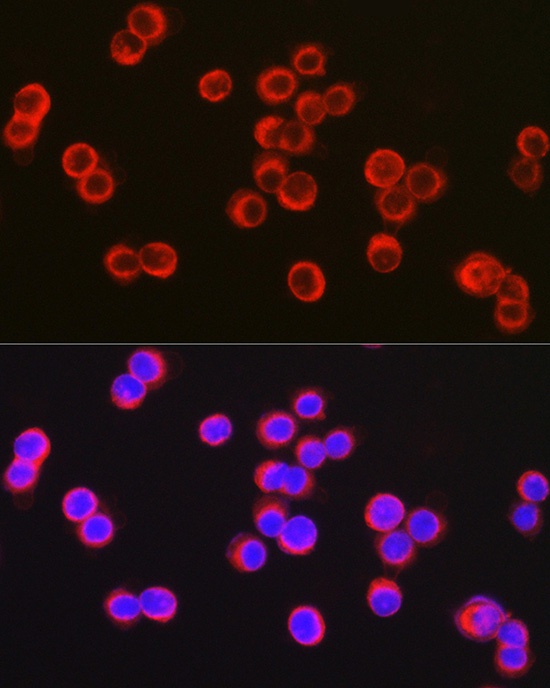
Immunofluorescence analysis of HT-29 cells using CFTR Rabbit pAb (CAB8386) at dilution of 1:100 (40x lens). Secondary antibody: Cy3-conjugated Goat anti-Rabbit IgG (H+L) (CABS007) at 1:500 dilution. Blue: DAPI for nuclear staining.