The Hemoglobin subunit alpha (HBA1) Monoclonal Antibody (CAB9293) is a high-quality antibody developed for reliable detection and analysis of target proteins. This antibody, developed using rabbit monoclonal technology, is highly specific and reactive with human samples, making it an ideal choice for a wide range of applications in biochemistry and molecular biology.The hemoglobin subunit alpha protein is a key component of hemoglobin, the molecule responsible for transporting oxygen in red blood cells. Studying this protein can provide valuable insights into conditions such as anemia, sickle cell disease, and other blood disorders. The CAB9293 antibody enables precise detection and analysis of the hemoglobin subunit alpha protein in various cell types, allowing researchers to delve deeper into its role in health and disease.
This antibody is validated for use in WB, IHC-P, ELISA, IF-P applications and has demonstrated reactivity against Human, Mouse, Rat samples.
The human alpha globin gene cluster located on chromosome 16 spans about 30 kb and includes seven loci: 5'- zeta - pseudozeta - mu - pseudoalpha-1 - alpha-2 - alpha-1 - theta - 3'. The alpha-2 (HBA2) and alpha-1 (HBA1) coding sequences are identical. These genes differ slightly over the 5' untranslated regions and the introns, but they differ significantly over the 3' untranslated regions. Two alpha chains plus two beta chains constitute HbA, which in normal adult life comprises about 97% of the total hemoglobin; alpha chains combine with delta chains to constitute HbA-2, which with HbF (fetal hemoglobin) makes up the remaining 3% of adult hemoglobin. Alpha thalassemias result from deletions of each of the alpha genes as well as deletions of both HBA2 and HBA1; some nondeletion alpha thalassemias have also been reported.
Purification Method
Affinity purification
Gene ID
3039
RRID
AB_2863705
Buffer Information
Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS containing 50% glycerol and 0.05% BSA, preserved with proclin300 or sodium azide, pH 7.3.
Western blot analysis of lysates from Rat liver using Hemoglobin subunit alpha (HBA1) Rabbit mAb (CAB9293) at 1:1000 dilution incubated overnight at 4℃. Secondary antibody: HRP-conjugated Goat anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25 μg per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (AbGn00020). Exposure time: 1s.
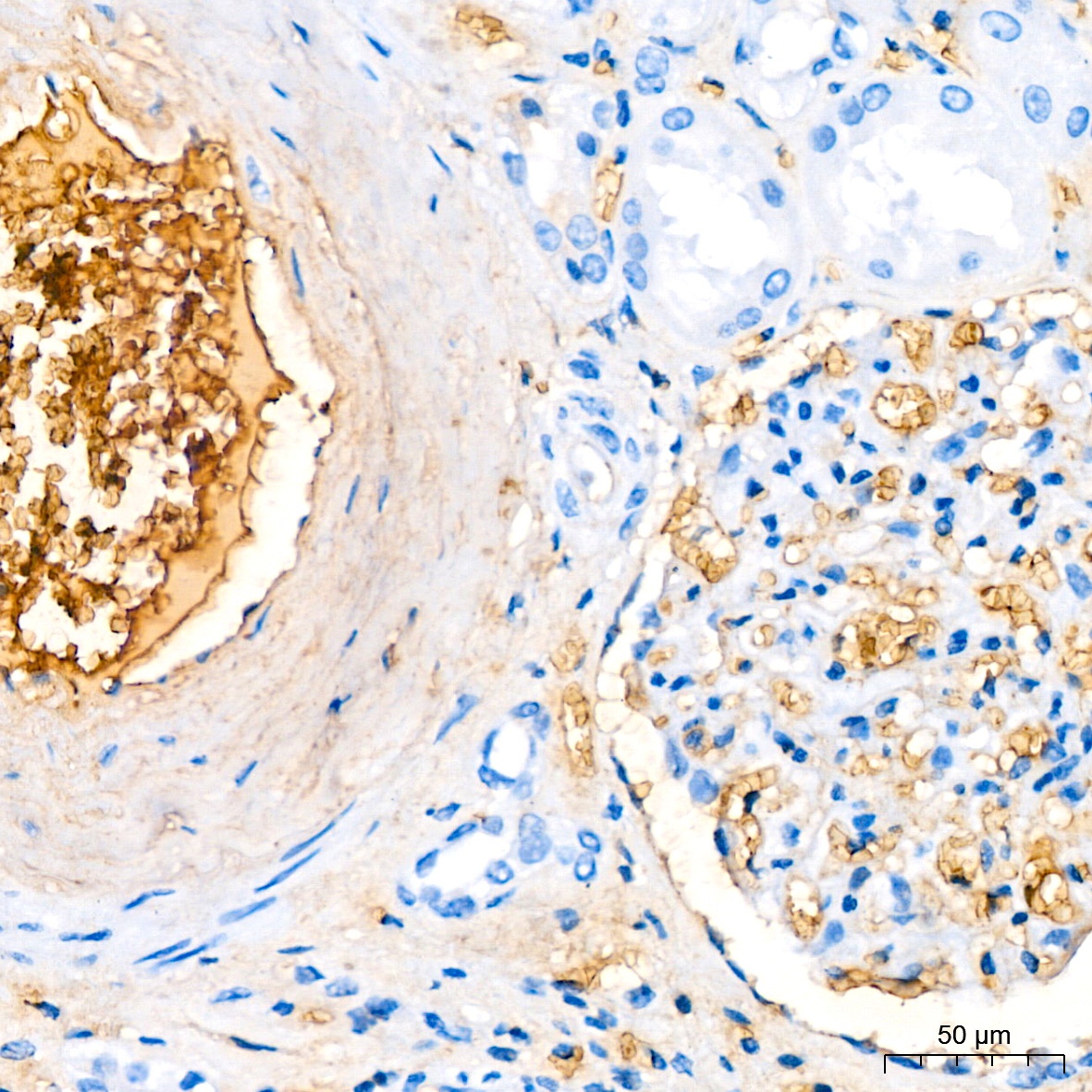
Immunohistochemistry analysis of paraffin-embedded Human kidney tissue using Hemoglobin subunit alpha (HBA1) Rabbit mAb (CAB9293) at a dilution of 1:200 (40x lens). High pressure antigen retrieval performed with 0.01M Citrate Buffer (pH 6.0) prior to IHC staining.
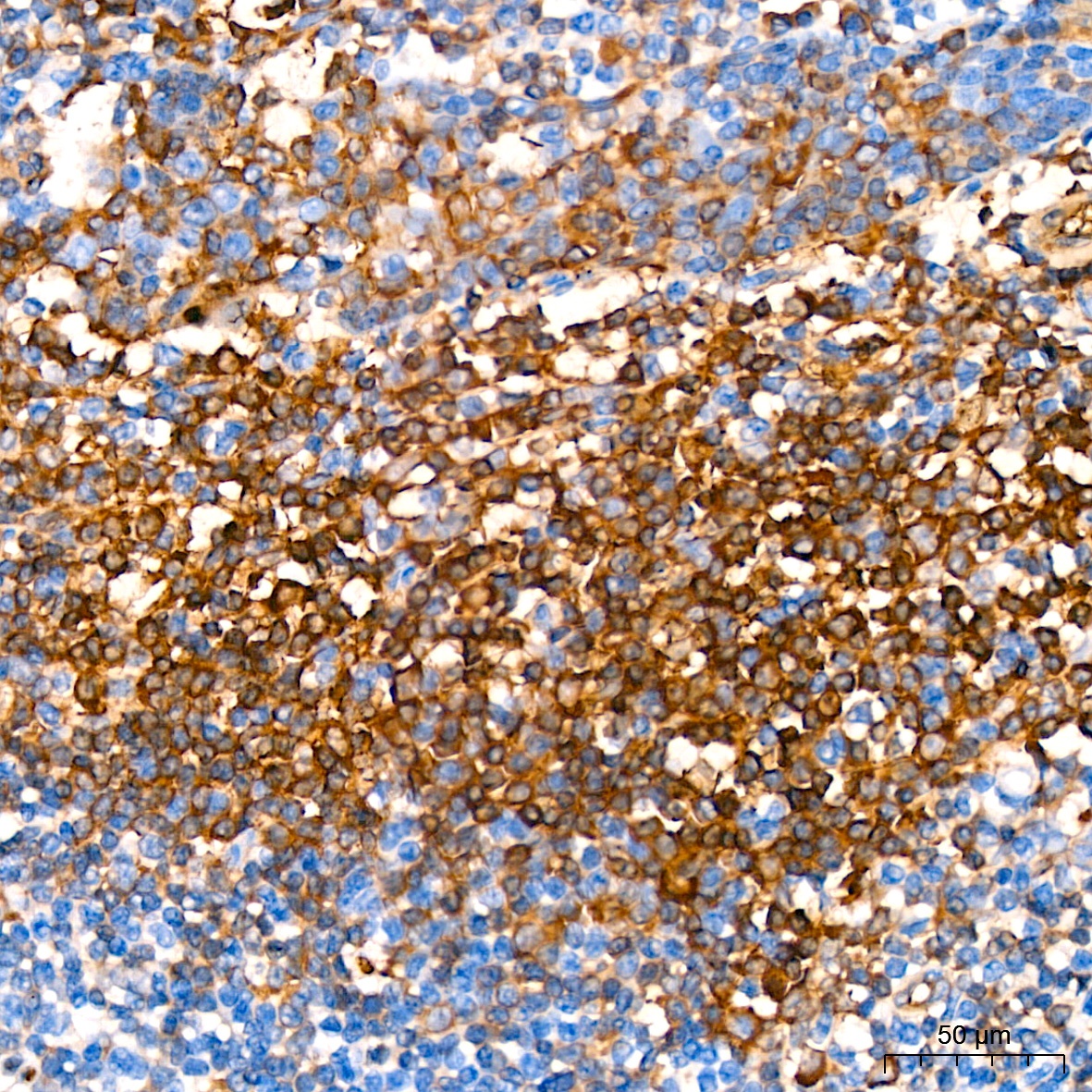
Immunohistochemistry analysis of paraffin-embedded Human tonsil tissue using Hemoglobin subunit alpha (HBA1) Rabbit mAb (CAB9293) at a dilution of 1:200 (40x lens). High pressure antigen retrieval performed with 0.01M Citrate Buffer (pH 6.0) prior to IHC staining.
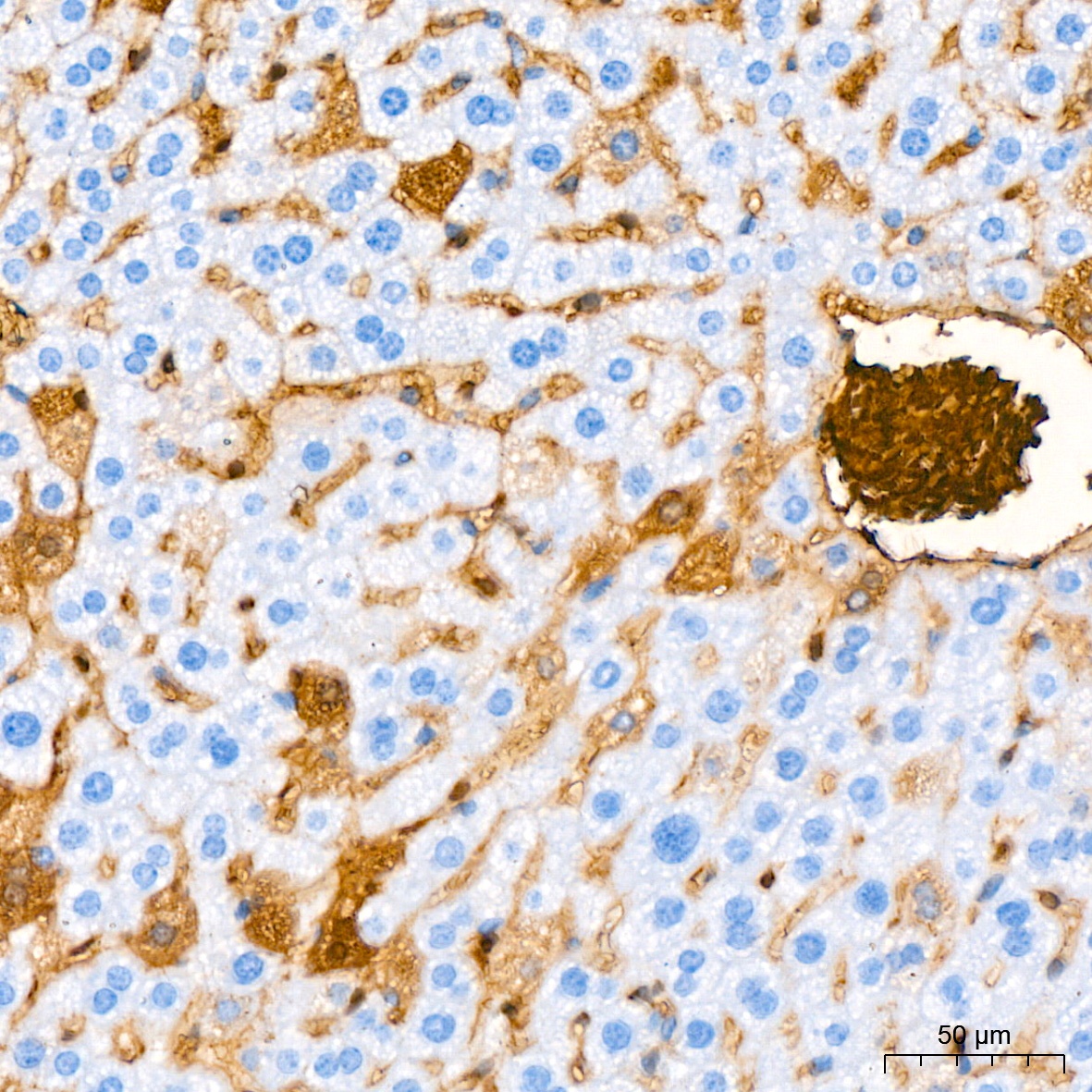
Immunohistochemistry analysis of paraffin-embedded Mouse liver tissue using Hemoglobin subunit alpha (HBA1) Rabbit mAb (CAB9293) at a dilution of 1:200 (40x lens). High pressure antigen retrieval performed with 0.01M Citrate Buffer (pH 6.0) prior to IHC staining.
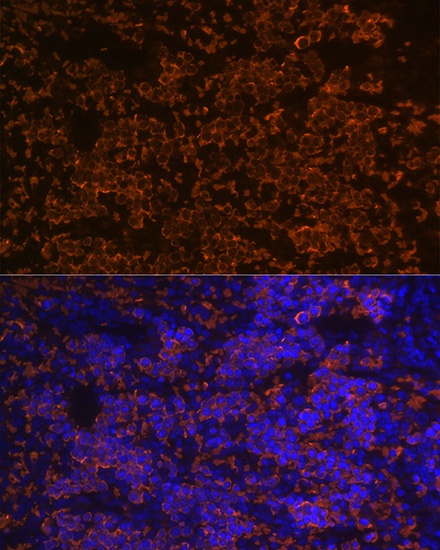
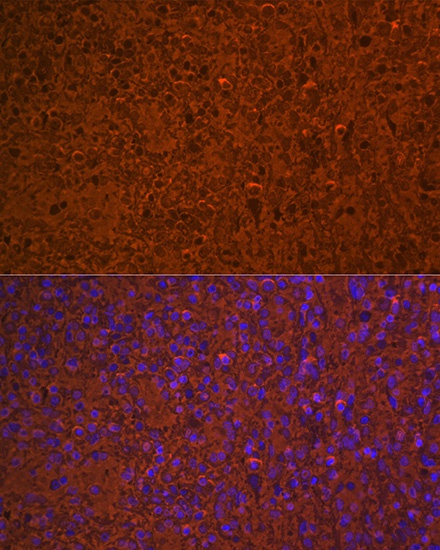
Immunofluorescence analysis of paraffin-embedded rat spleen using Hemoglobin subunit alpha (HBA1) Rabbit mAb (CAB9293) at dilution of 1:100 (40x lens). Secondary antibody: Cy3-conjugated Goat anti-Rabbit IgG (H+L) (CABS007) at 1:500 dilution. Blue: DAPI for nuclear staining.
Immunofluorescence analysis of paraffin-embedded human spleen using Hemoglobin subunit alpha (HBA1) Rabbit mAb (CAB9293) at dilution of 1:100 (40x lens). Secondary antibody: Cy3-conjugated Goat anti-Rabbit IgG (H+L) (CABS007) at 1:500 dilution. Blue: DAPI for nuclear staining.




 ELISA Kit (HUEB0705)")

")
![Anti-Hemoglobin subunit alpha [R06-5F7] Monoclonal Antibody (AGMB01073)](https://cdn11.bigcommerce.com/s-h68l9z2lnx/images/stencil/590x590/products/272362/693147/anti-hemoglobin-subunit-alpha-r06-5f7-monoclonal-antibody-agmb01073__77270.1774508374.jpg?c=2 "Anti-Hemoglobin subunit alpha [R06-5F7] Monoclonal Antibody (AGMB01073)")
 ELISA Kit (RTEB0313)")