
Waardenburg Syndrome and Klein-Waardenburg Syndrome
Waardenburg Syndrome: A Rare Genetic Disorder Affecting Pigmentation, Hearing, and More
Waardenburg Syndrome (WS) is a fascinating and rare genetic disorder that impacts various aspects of an individual's appearance and sensory abilities. First described by Dutch ophthalmologist Petrus Johannes Waardenburg in the 1950s, WS is characterized by distinctive features, including changes in pigmentation, hearing loss, and facial abnormalities.
Table of Contents
Jump to a section:
What is Waaredenburg Syndrome?
Waardenburg Syndrome (WS) is a genetically heterogeneous disorder which is characterized by a distinct fusion of features, including changes in pigmentation, hearing loss, and facial abnormalities. The condition arises from mutations in specific genes critical for the development and function of melanocytes and other neural crest-derived cells.
Waardenburg Syndrome is considered a rare genetic disorder, and its prevalence varies among different populations. The estimated prevalence of Waardenburg Syndrome is approximately 1 in 42,000 to 1 in 50,000 individuals worldwide. However, the prevalence can be higher in certain populations or regions.
Classification of Waardenburg Syndrome
Waredenburg Syndrom is classified into four primary types, each with its own unique features and genetic basis. The four primary types of Waardenburg Syndrome are designated as Type 1 (WS1), Type 2 (WS2), Type 3 (WS3), and Type 4 (WS4).
-
Waardenburg Syndrome Type 1 (WS1):
WS1 is associated with mutations in the PAX3 gene, leading to disturbances in neural crest cell migration and development. The defining feature of WS1 is dystopia canthorum, characterized by widely spaced inner corners of the eyes. Individuals with WS1 may also present with pigmentation changes in the eyes, hair, and skin, sensorineural hearing loss, and a predisposition to neural tube defects. -
Waardenburg Syndrome Type 2 (WS2):
WS2 is primarily caused by mutations in the MITF (Microphthalmia-associated transcription factor) gene. WS2 lacks dystopia canthorum, differentiating it from WS1. Instead, individuals with WS2 may exhibit a broader spectrum of pigmentation variations, including heterochromia iridum (eyes of different colors) and a white forelock. Sensorineural hearing loss is another consistent feature in WS2. -
Waardenburg Syndrome Type 3 (WS3) - Klein-Waardenburg Syndrome:
WS3 is distinguished by its characteristic combination of features, including pigmentary changes in the eyes and hair, sensorineural hearing loss, and limb abnormalities. WS3 is unique due to the presence of limb defects, often involving hypoplastic or absent thumbs and forearm abnormalities. -
Waardenburg Syndrome Type 4 (WS4) - Waardenburg-Shah Syndrome:
WS4 encompasses individuals with features of both Waardenburg Syndrome and Hirschsprung disease, a disorder characterized by the absence of ganglion cells in the colon. WS4 is characterized by pigmentation abnormalities, sensorineural hearing loss, Hirschsprung disease, and occasionally other neural crest-related anomalies.
Causes of Waardenburg Syndrome
Waardenburg Syndrome is caused by mutations in genes that influence the functioning of neural crest cells during early embryonic development. These neural crest cells are migratory cells that give rise to various tissues and structures in the body, including the eyes, ears, skin, and facial bones.
The specific genes responsible for different types of Waardenburg Syndrome are as follows:
Type 1 (WS1): Mutations in the PAX3 gene on chromosome 2q36.1 are commonly associated with WS1.
Type 2 (WS2): WS2 can be caused by mutations in genes such as MITF on chromosome 3p14.1-p12.3.
Type 2A (WS2A): WS2A is linked to mutations in the PAX3 gene, similar to WS1.
Type 2B (WS2B): WS2B is caused by mutations in the SNAI2 (Slug) gene on chromosome 8q11.21.
Type 2C (WS2C): WS2C is associated with mutations in the SOX10 gene on chromosome 22q13.1.
Type 2D (WS2D): WS2D is caused by mutations in the endothelin receptor type B (EDNRB) gene on chromosome 13q22.
Type 3 (WS3): WS3 is primarily associated with mutations in the PAX3 gene, similar to WS1.
Type 4 (WS4): WS4 is genetically heterogeneous, with some cases attributed to mutations in the EDNRB (Endothelin receptor type B) gene on chromosome 13q22, whereas others result from mutations in the EDN3 (Endothelin-3) gene on chromosome 20q13.12.
Type 4A (WS4A): Caused by mutations in the EDNRB gene on chromosome 13q22.
Type 4B (WS4B): Caused by mutations in the EDN3 gene on chromosome 20q13.12.
Type 4C (WS4C): Caused by mutations in the SOX10 gene on chromosome 22q13.1.
Waardenburg Syndrome is typically inherited in an autosomal dominant pattern, where a person only needs to inherit one copy of the mutated gene from one parent to express the syndrome. However, some rare cases may follow an autosomal recessive pattern, where an individual needs to inherit two copies of the mutated gene, one from each parent.
Waardenburg Syndrome Inheritance Pattern. WS is usually inherited in an autosomal-dominant pattern. However, type 2D, 3, 4A, and 4B can have an autosomal-recessive pattern.
Summary of Classification
| WS Type | Gene | Inheritance Pattern |
|
Type 1 (WS1) |
PAX3 (chromosome 2q36.1) |
Autosomal Dominant |
|
Type 2A (WS2A) |
PAX3 (chromosome 2q36.1) |
Autosomal Dominant |
|
Type 2B (WS2B) |
SNAI2 (Slug) (chromosome 8q11.21) |
Autosomal Dominant |
|
Type 2C (WS2C) |
SOX10 (chromosome 22q13.1) |
Autosomal Dominant |
|
Type 2D (WS2D) |
EDNRB (chromosome 13q22) |
Autosomal Recessive |
|
Type 3 (WS3) |
PAX3 (chromosome 2q36.1) |
Autosomal Dominant |
|
Type 4A (WS4A) |
EDNRB (chromosome 13q22) |
Autosomal Dominant / Recessive |
|
Type 4B (WS4B) |
EDN3 (chromosome 20q13.12) |
Autosomal Dominant / Recessive |
|
Type 4B (WS4B) |
SOX10 (chromosome 22q13.1) |
Autosomal Recessive |
Waardenburg Syndrome Type 3 (WS3)
Waardenburg Syndrome Type 3, also known as Klein-Waardenburg Syndrome, presents a unique combination of clinical features that distinguish it from other types of Waardenburg Syndrome. While WS3 shares some characteristics with WS1 and WS2, it is the presence of limb abnormalities that sets it apart.
Characteristics of Waardenburg Syndrome Type 3
-
Pigmentation Abnormalities:
Individuals with WS3 often exhibit distinctive pigmentation changes affecting the eyes, hair, and skin. Eye abnormalities may include strikingly vibrant blue eyes, heterochromia iridum (eyes of different colors), or a pale blue eye color. Moreover, a white forelock or premature graying of the hair is a common feature. -
Sensorineural Hearing Loss:
Sensorineural hearing loss, caused by abnormalities in the inner ear or auditory nerve, is a hallmark feature of WS3. The severity of hearing impairment may vary, ranging from mild to profound. -
Limb Abnormalities:
The most distinguishing feature of WS3 is the presence of limb defects, predominantly affecting the upper extremities. Individuals may have hypoplastic (underdeveloped) or absent thumbs and forearm abnormalities. These limb anomalies are a result of disruptions in the development of neural crest-derived structures. -
Facial Characteristics:
Similar to other types of Waardenburg Syndrome, individuals with WS3 may have facial features such as a wide nasal bridge and dystopia canthorum, a condition where the inner corners of the eyes are widely spaced. -
Possible Neural Tube Defects:
In some cases, individuals with WS3 may have an increased risk of neural tube defects, such as spina bifida or other abnormalities in the development of the spinal cord.
Diagnosis of Waardenburg Syndrome Type 3
Diagnosing Waardenburg Syndrome Type 3 requires a comprehensive evaluation, including clinical assessment of physical features and family history. Audiological evaluations are crucial to identify sensorineural hearing loss. Genetic testing confirms PAX3 gene mutations associated with WS3, ensuring an accurate diagnosis and enabling appropriate management and genetic counseling.
Waaredenburg Syndrome Related Kits
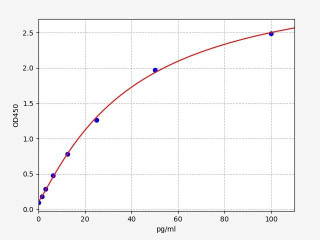
| Human Endothelin-3 / EDN3 ELISA Kit | |
|---|---|
| ELISA Type | Sandwich |
| Sensitivity | 0.938pg/ml |
| Range | 1.563-100pg/ml |
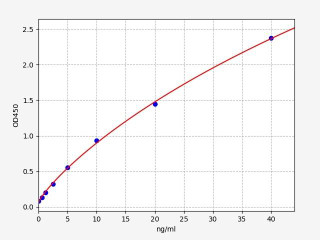
| Human MITF ELISA Kit | |
|---|---|
| ELISA Type | Sandwich |
| Sensitivity | 0.375ng/ml0.375ng/ml0.375ng/ml |
| Range | 0.625-40ng/ml |
Written by Lauryn McLoughlin
Lauryn McLoughlin completed her undergraduate degree in Neuroscience before completing her masters in Biotechnology at University College Dublin.
Recent Posts
-
Introducing BioSymposia: The Gathering Place for Science
Announcement · Powered by Assay GenieIntroducing BioSymposia: The Gathering Place for ScienceWe’re e …21st Jul 2026 -
ELISA vs Western Blot: Which Technique Should You Choose?
Written by Seán Mac Fhearraigh, PhD • Updated: 19 May 2026 • ~9 min read Quick Answer ELISA …20th May 2026 -
Types of ELISA: Direct, Indirect, Sandwich & Competitive Compared
Written by Seán Mac Fhearraigh, PhD • Updated: 19 May 2026 • ~9 min read Quick Answer The fi …20th May 2026