
Types of PCR – 7 Methods, Principles & Applications
Types of PCR – 7 Methods, Principles & Applications
PCR stands as a cornerstone in molecular biology, enabling scientists to magnify and analyze fragments of DNA with incredible precision — from standard amplification to RT-PCR, qPCR, digital PCR and beyond, alongside the high-fidelity polymerases and master mixes to run them.
Browse Molecular Biology Tools →PCR stands as a cornerstone in molecular biology, enabling scientists to magnify and analyze fragments of DNA with incredible precision. In this blog, we'll embark on understanding of PCR – from its fundamental principles to the diverse techniques that have revolutionized research, diagnostics, and beyond.
Key Takeaways
- PCR is a pivotal technique for DNA amplification, crucial in research, diagnostics, and forensics.
- Variants like Hot Start PCR, RT-PCR, and qPCR enhance specificity, analyze RNA, and quantify nucleic acids.
- Advances like High Fidelity Polymerase and Digital PCR offer precision in detecting mutations and low-abundance genes.
Table of Contents
Jump to a section:
PCR reagents & molecular biology kits
Assay Genie supplies high-fidelity polymerases, master mixes, cloning kits and mycoplasma detection assays to run every stage of your PCR and molecular biology workflow.

Genie Fusion Ultra High-Fidelity 2X Master Mix
High-fidelity 2X master mix for accurate, robust PCR amplification and cloning.
View product
Genie Fusion Ultra High-Fidelity Red 2X Master Mix
Dye-loaded high-fidelity master mix for direct gel loading after PCR.
View product
GenieClone DNA Assembly Cloning Kit
Seamless, sequence-independent assembly of PCR fragments into any vector.
View kit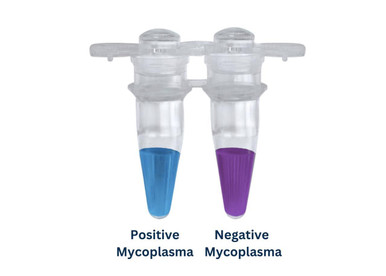
MycoGenie Rapid Mycoplasma Detection Kit
PCR-based rapid detection of mycoplasma contamination in cell cultures.
View kit
GenieColor Mycoplasma Detection Kit
Visual pink-to-yellow colour change detects mycoplasma in 1 hour, no instrument needed.
View kitPCR
Polymerase Chain Reaction (PCR) is a powerful laboratory technique used to amplify specific segments of DNA, creating millions of copies from a single piece of DNA. Developed in the 1980s, PCR has become an essential tool in various fields of biological research, diagnostics, forensics, and more. It revolutionized DNA analysis by allowing scientists to create sufficient amounts of DNA for analysis, even from limited or degraded samples.
The PCR process involves a series of temperature-dependent steps and utilizes a DNA polymerase enzyme to catalyze the synthesis of new DNA strands. Here's a step-by-step explanation of the PCR process:
- Denaturation (94-98°C): The double-stranded DNA template is heated to a high temperature, usually around 94-98°C. This causes the hydrogen bonds between the complementary DNA strands to break, resulting in the separation of the two strands. This step is often referred to as "melting."
- Annealing (50-65°C): The temperature is lowered to a specific range (typically 50-65°C), allowing short DNA primers to bind to the complementary sequences at the beginning of the target DNA region. These primers serve as starting points for DNA synthesis.
- Extension (72°C): The temperature is raised to around 72°C, which is the optimal temperature for the DNA polymerase enzyme (usually Taq polymerase) to synthesize a new DNA strand by adding nucleotides complementary to the template strand. The primers dictate the sequence of the newly synthesized DNA strand.
These three steps—denaturation, annealing, and extension—are typically repeated for a specific number of cycles, usually around 20 to 40 cycles. Each cycle doubles the amount of DNA, resulting in an exponential increase in the target DNA segment.
Fig: Polymerase Chain Reaction (PCR)
Standard PCR techniques face a hurdle when dealing with guanine/cytosine-rich (GC-rich) areas. Sequences with more GC are more stable than those with less GC. Additionally, GC-rich sequences are more likely to generate secondary structures like hairpin loops. As a result, during the denaturation step, GC-rich double strands are challenging to fully separate. As a result, DNA polymerase encounters difficulties creating the new strand. This can be made better by changes to a higher denaturation temperature and shorter annealing times, which also help to prevent the unspecific binding of GC-rich primers. The amplification of GC-rich sequences can be improved by the addition of additional reagents. As a result of the disruption of the secondary structures brought on by GC interactions, the separation of the double strands is facilitated by the application of DMSO, glycerol, and betaine.
Hot Start PCR
Hot-Start PCR is a modified version of the traditional polymerase chain reaction (PCR) technique that is designed to enhance the specificity and efficiency of DNA amplification. It addresses a common issue in standard PCR, where non-specific amplification can occur due to the premature activation of the DNA polymerase enzyme during the initial stages of the reaction setup. This can lead to the generation of undesired products and reduced overall specificity. In Hot-Start PCR, measures are taken to prevent the DNA polymerase from being active at low temperatures, such as during the initial setup and the initial heating steps of the PCR process. The goal is to prevent the amplification of non-specific products and enhance the amplification of the intended target sequence. This is particularly valuable when working with complex DNA templates or when dealing with samples that contain trace amounts of target DNA. The advantages of Hot-Start PCR include enhanced specificity, increased sensitivity and reduced optimization.
There are several methods used to implement the Hot-Start PCR technique:
- Physical Separation: One common approach involves physically separating the DNA polymerase from its DNA template and primers during the initial setup. This separation is achieved by using modified DNA polymerases or antibody-based techniques. For example, an antibody can be used to inhibit the polymerase's activity until the reaction is heated, at which point the antibody denatures, allowing the polymerase to become active.
- Chemical Modification: Another method involves chemically modifying the DNA polymerase enzyme itself. Chemical modifications are added to the enzyme that render it inactive at lower temperatures. As the reaction temperature increases during the initial heating steps, these modifications are removed, activating the enzyme for DNA amplification.
- Hot-Start Taq Polymerase: Some commercial DNA polymerases are designed to be inactive at room temperature or lower, but become active at elevated temperatures. These specialized enzymes, often referred to as "Hot-Start Taq Polymerases," possess antibody-mediated inhibition, reversible chemical modifications, or other mechanisms to prevent their activity until the reaction is properly heated.
High Fidelity Polymerase
Nucleotide matching errors can happen even though DNA polymerases amplify to the original template sequence quite precisely. In applications like cloning and genome editing, mismatches can lead to shortened transcripts and proteins that have been incorrectly translated or are inactive. Polymerases with a "proofreading" activity have been found and incorporated into the workflow to prevent these mismatches. In Pyrococcus furiosus, the first proofreading polymerase, Pfu, was discovered in 1991. The 3' to 5' exonuclease activity of this Pfu enzyme. The exonuclease eliminates mismatched nucleotides at the 3' end of the strand as the DNA is amplified. The proper nucleotide is then substituted, and DNA synthesis keeps going.
The proper nucleoside triphosphate's binding affinity with the enzyme, where ineffective binding delays synthesis and permits the right replacement, is used to identify erroneous nucleotide sequences. Compared to Taq DNA polymerase, Pfu polymerase's proofreading activity leads to fewer errors in the final sequence. In order to further lower the mistake rate during DNA amplification, additional proofreading enzymes have been discovered recently, and the original Pfu enzyme has been modified.
RT-PCR
Reverse Transcription Polymerase Chain Reaction, commonly known as RT-PCR, is a powerful molecular biology technique that combines the principles of reverse transcription and polymerase chain reaction. This method is specifically designed to analyze and amplify RNA molecules, converting them into complementary DNA (cDNA) for further analysis.
The process begins with the reverse transcription step, where an enzyme called reverse transcriptase synthesizes a complementary DNA strand (cDNA) using a single-stranded RNA molecule as a template. This step is crucial because many biological processes involve RNA, such as gene expression and viral replication, and converting RNA to cDNA allows researchers to study these processes more easily.
Once the cDNA is generated, the polymerase chain reaction is employed to amplify specific target sequences. This involves repeated cycles of heating and cooling the reaction mixture. During the heating step (denaturation), the DNA strands are separated, creating single-stranded templates. In the cooling step (annealing), short DNA primers specifically designed to bind to the target cDNA sequences attach to the templates. Then, a heat-stable DNA polymerase enzyme extends the primers by adding complementary nucleotides, resulting in the synthesis of new DNA strands.
The end result of RT-PCR is an amplified amount of cDNA from the original RNA sample, allowing researchers to study gene expression levels, detect viral infections, analyze RNA sequences, and more. RT-PCR has proven invaluable in various fields, from medical diagnostics, where it's used to detect diseases like COVID-19, to molecular biology research, where it enables the exploration of gene function and regulation. Its ability to convert RNA into DNA and then amplify specific sequences has made RT-PCR an indispensable tool in modern molecular biology.
qPCR and RT-qPCR
For many applications, nucleic acids are detected, described, and quantified using quantitative PCR (qPCR). In RT-qPCR, RNA transcripts are frequently quantified by first reverse transcribed into cDNA, as previously mentioned, and then qPCR is performed. Denaturation, annealing, and elongation are three processes that are repeated to amplify DNA, much like in conventional PCR. But in qPCR, fluorescent labeling makes it possible to gather data as the PCR proceeds. Due to the variety of techniques and chemistries accessible, this technology has several advantages.
Fluorescent labeling, which uses a dsDNA binding dye, enables the measurement of the amplified DNA molecules in dye-based qPCR (usually green). The fluorescence is gauged over each cycle. The amount of DNA that has been duplicated causes the fluorescence signal to rise correspondingly. As a result, the DNA is measured "real-time". Only one target may be analyzed at a time with dye-based qPCR, and any ds-DNA found in the sample will cause the dye to bind.
Many targets can be identified simultaneously in each sample using probe-based qPCR, but this needs the creation and development of a target-specific probe or probes, which are employed in addition to primers. There are many other sorts of probe designs, but the most popular kind is a hydrolysis probe that combines a fluorophore with a quencher. When the probe is still intact, fluorescence resonance energy transfer (FRET) stops the fluorophore from emitting through the quencher. However, the probe is hydrolyzed during primer extension and amplification of the particular sequence it is linked to during the PCR reaction. An amplification-dependent rise in fluorescence is produced as a result of the probe's cleavage, which frees the fluorophore from the quencher.
As a result, the amount of the probe target sequence present in the sample directly correlates with the fluorescence signal from a probe-based qPCR process. In qPCR-based diagnostic tests, probe-based qPCR is frequently employed because it is more precise than dye-based qPCR.
Fig: Steps in qPCR
Isothermal Amplification
For the PCR methods discussed above to precisely ramp up and down chamber temperatures for the denaturation, annealing, and extension phases, expensive thermocycling equipment is needed. Numerous methods have been devised that don't require such exact equipment and can be used inside the target cells or even in a straightforward water bath. These methods, which are referred to as isothermal amplification collectively, operate on the principles of exponential, linear, or cascade amplification.
Loop-mediated isothermal amplification, or LAMP, is the most well-known kind of isothermal amplification. LAMP amplifies template DNA or RNA using exponential amplification at 650C. Using a DNA polymerase and four to six primers that are complementary to specific sections of the target DNA, LAMP creates new DNA. A "loop" structure might form in the freshly synthesized DNA as a result of two of these primers' complementary sequences recognizing and binding to sequences in the other primers. This structure then facilitates primer annealing in subsequent rounds of amplification. Numerous techniques, such as fluorescence, agarose gel electrophoresis, or colorimetry, can be used to see LAMP.
LAMP was a suitable alternative for SARS-CoV-2 testing in locations where clinical lab testing was not easily accessible, where sample storage and transport was not practical, or in labs that did not previously have PCR thermocycling equipment because it was simple to visualize and detect the presence or absence of product by colorimetry and did not require expensive equipment.
Digital PCR
Digital Polymerase Chain Reaction, or Digital PCR, is a cutting-edge molecular biology technique that takes the principles of traditional PCR to a new level of precision and sensitivity. Digital PCR is used to accurately quantify and analyze DNA or RNA molecules present in a sample, even when they are present in very low concentrations.
In Digital PCR, the sample is partitioned into thousands of individual reactions, each containing a single molecule or a few molecules of the target DNA or RNA. This partitioning is achieved using microfluidic devices or specialized emulsion techniques. Each partitioned reaction acts as a miniature PCR reaction, with either the target DNA/RNA amplifying or remaining unamplified, depending on its presence or absence.
After amplification, the partitions are analyzed, and the number of positive partitions (those where amplification occurred) and negative partitions (those without amplification) is counted. This information is used to calculate the absolute quantity of the target molecules in the original sample. Digital PCR's ability to detect and quantify rare sequences with high precision makes it particularly valuable for applications such as detecting genetic mutations, studying gene expression at low levels, and analyzing complex mixtures of DNA or RNA — a capability increasingly relevant to liquid biopsy and ctDNA analysis.
Compared to traditional quantitative PCR (qPCR), Digital PCR offers several advantages, including enhanced sensitivity, reduced susceptibility to PCR inhibitors, and improved accuracy for low-abundance targets. It's a powerful tool in various fields, including medical diagnostics, genetic testing, environmental monitoring, and more, where accurate quantification of nucleic acids is crucial. By providing a digital, absolute quantification of target molecules, Digital PCR contributes to more reliable and precise molecular analyses.
What Is PCR and How Does It Work?
Polymerase Chain Reaction (PCR) is a molecular biology technique that exponentially amplifies specific DNA sequences. The process occurs in repeated cycles of three temperature-dependent steps: (1) Denaturation at 94–95°C separates double-stranded DNA; (2) Annealing at 50–65°C allows primers to bind; (3) Extension at 72°C uses Taq polymerase to synthesize new strands. Over 25–35 cycles, this produces millions to billions of copies of the target sequence.
What Are the Different Types of PCR?
| Type | Principle | Key Application | Sensitivity |
|---|---|---|---|
| Standard PCR | End-point amplification | Gene cloning, pathogen screening | High (~1 pg DNA) |
| RT-PCR | Reverse transcriptase converts RNA to cDNA | RNA detection, viral RNA | High (1–10 copies) |
| qPCR (Real-time) | Fluorescent reporter monitored cycle-by-cycle | Quantification, viral load | Very high (0.1–1 copy) |
| RT-qPCR | RT + real-time monitoring | COVID-19 diagnostics, gene expression | Very high (1 copy RNA) |
| Digital PCR | Partitioned reactions; Poisson statistics | Rare mutation detection | Single-molecule |
| Hot-Start PCR | Antibody blocks polymerase until 95°C | Improved specificity | High |
| Nested PCR | Two sequential amplifications | Ultra-low copy detection | Extreme |
| Multiplex PCR | Multiple primer pairs in single reaction | Viral panels, SNP genotyping | Good |
What Is the Difference Between RT-PCR and qPCR?
| Feature | RT-PCR | qPCR | RT-qPCR |
|---|---|---|---|
| Target | RNA (converted to cDNA) | DNA | RNA (cDNA in real-time) |
| Detection | End-point; gel electrophoresis | Real-time; fluorescence each cycle | Real-time; fluorescence each cycle |
| Quantification | Semi-quantitative | Quantitative (Ct/Cq value) | Quantitative (Ct/Cq value) |
| Speed | 2–3 hours | 45–90 min | 45–90 min |
| Common Use | Gene expression screening | Viral load, pathogen detection | COVID-19 diagnostics |
What Is Digital PCR and How Does It Compare to qPCR?
Digital PCR (dPCR) partitions the sample into thousands to millions of independent microdroplets, each assessed as positive or negative after amplification. Quantification relies on Poisson statistics rather than fluorescence kinetics, enabling absolute quantification without external standards. dPCR provides single-molecule sensitivity and is superior for detecting rare mutations (0.01% sensitivity) and accurate copy number variation analysis. However, dPCR requires specialized equipment and costs more per sample. qPCR remains faster and sufficient for most applications.
How Do You Choose the Right Type of PCR?
- Step 1: Target nucleic acid? RNA → RT-PCR or RT-qPCR. DNA → Step 2.
- Step 2: Quantification needed? Yes → qPCR or dPCR. Qualitative only → standard or nested PCR.
- Step 3: Sensitivity? Routine (>100 copies) → standard PCR. Sensitive (1–100 copies) → qPCR. Ultra-sensitive → dPCR or nested PCR.
- Step 4: Multiple targets? Yes → multiplex PCR. Single target → standard formats.
- Step 5: Cost/time? Budget-limited → standard PCR. Rapid turnaround → qPCR (1.5–2 hours). Maximum accuracy → dPCR.
Setting up your next PCR experiment?
From ultra high-fidelity master mixes to cloning kits and mycoplasma detection, Assay Genie's molecular biology tools give you reliable, publication-ready results — with fast delivery and expert PhD-level technical support.
Explore Molecular Biology Tools →Frequently Asked Questions About PCR Types
What is the difference between PCR and RT-PCR?
Standard PCR amplifies DNA, while RT-PCR amplifies RNA by first converting it to cDNA using reverse transcriptase. RT-PCR is essential for detecting viral RNA (SARS-CoV-2, influenza) and quantifying mRNA expression.
Which type of PCR is most sensitive?
Digital PCR (dPCR) offers the highest sensitivity, detecting single molecules. qPCR and RT-qPCR follow closely at 0.1–1 copy per reaction. Nested PCR also approaches single-molecule sensitivity.
What is hot-start PCR used for?
Hot-start PCR uses temperature-sensitive polymerase to prevent non-specific amplification during setup, only activating at 95°C. This improves specificity and reduces primer-dimer formation.
Can you use PCR to detect RNA viruses?
Yes, but RNA viruses require RT-PCR or RT-qPCR, which first convert viral RNA into cDNA. RT-qPCR is the gold standard for detecting SARS-CoV-2, influenza, dengue, and Ebola.
What is the difference between endpoint PCR and real-time PCR?
Endpoint PCR detects product after all cycles via gel electrophoresis (qualitative). Real-time PCR monitors product accumulation each cycle via fluorescence (quantitative, faster, more sensitive).
In conclusion, this exploration into Polymerase Chain Reaction (PCR) and its diverse types underscores the paramount significance of this revolutionary molecular biology technique. By adeptly amplifying and analyzing nucleic acids, PCR techniques enable us to unravel intricate genetic information and unravel previously inaccessible insights. Our investigation has delved into the intricacies of PCR variants, encompassing Reverse Transcription PCR (RT-PCR) and their multifaceted applications. The elucidation of these methodologies and their inherent capabilities serves to underscore the crucial role they play in modern molecular biology research and diagnostic
Related articles
Recent Posts
-
ELISA vs Western Blot: Which Technique Should You Choose?
Written by Seán Mac Fhearraigh, PhD • Updated: 19 May 2026 • ~9 min read Quick Answer ELISA …20th May 2026 -
Types of ELISA: Direct, Indirect, Sandwich & Competitive Compared
Written by Seán Mac Fhearraigh, PhD • Updated: 19 May 2026 • ~9 min read Quick Answer The fi …20th May 2026 -
Sandwich ELISA: Step-by-Step Protocol & Troubleshooting
Written by Seán Mac Fhearraigh, PhD • Updated: 19 May 2026 • ~ …20th May 2026