Apoptosis Unveiled: Your Complete Guide to Intrinsic & Extrinsic Pathways
Apoptosis: Your Complete Guide to Intrinsic & Extrinsic Pathways
Apoptosis is programmed cell death — the controlled, non-inflammatory removal of damaged or unwanted cells that is essential for development, tissue homeostasis and cancer prevention. This guide covers what apoptosis is, how it differs from necrosis, the intrinsic (mitochondrial) and extrinsic (death-receptor) pathways, the Bcl-2 family and caspases that drive them, and how apoptosis is measured.
Browse ELISA Kits →Key Takeaways
- Apoptosis is a programmed cell death process, vital for tissue maintenance and disease prevention.
- Distinct from necrosis, apoptosis involves cellular shrinkage, nuclear breakdown, membrane disruption, and fragmentation into apoptotic bodies.
- p53 pathway, intrinsic and extrinsic pathways, and caspase cascades are key mechanisms in apoptosis.
- Apoptosis vs. Necrosis: Apoptosis is controlled and natural, while necrosis is uncontrolled and harmful.
- p53 Pathway: Activated by DNA damage, involves proteins like Bax, Bak, Bad.
- Intrinsic Apoptosis: Triggered internally by cellular stress; extrinsic by external signals.
- Caspases: Crucial enzymes for apoptosis execution, grouped by function.
What is Apoptosis
- The cell shrinks and becomes rounder
- The cell's nucleus breaks down into fragments
- The cells' plasma membrane breaks down
- The cell breaks apart into smaller pieces (apoptotic bodies)

Apoptosis vs Necrosis
The main difference between apoptosis and necrosis is that apoptosis is a natural and controlled process of cell death while necrosis is an uncontrolled process of cell death. Apoptosis is important for the development and maintenance of healthy tissues while necrosis leads to tissue damage.
Apoptosis is a type of programmed cell death that occurs in response to various stimuli, such as DNA damage or viral infection. Apoptosis is characterized by several morphological changes, including cell shrinkage, plasma membrane bulging (known as blebbing), chromatin condensation, and formation of apoptotic bodies.
Necrosis, on the other hand, is a type of cell death that occurs in response to extreme stress, such as exposure to toxins or infection. Necrosis is characterized by cell swelling, membrane rupture, and release of inflammatory mediator.

p53 Apoptosis Pathway
As one of the most important tumor suppressor proteins, p53 is frequently mutated or lost in cancer cells. This can lead to uncontrolled cell growth and eventually tumor formation. p53 also plays a key role in apoptosis, or programmed cell death. When DNA is damaged, p53 is activated and triggers apoptosis to prevent the damaged cells from dividing and spreading.
There are several signaling proteins involved in p53-mediated apoptosis, including Bax, Bak, and Bad. Bax, Bak, and Bad are pro-apoptotic proteins that promote cell death. When DNA damage occurs and p53 is activated, Bax and Bak are recruited to the mitochondria, where they cause the release of cytochrome c. Cytochrome c triggers a cascade of events that leads to cell death. Bad is also phosphorylated by p53, which prevents it from inhibiting cell death.
In summary, p53 plays a vital role in mediating apoptosis in response to DNA damage. The p53 apoptosis pathway involves several key signaling proteins, including Bax, Bak, and Bad. When DNA damage occurs, p53 activates these proteins to trigger cell death and prevent the damaged cells from spreading.
Apoptosis Assays & ELISA Kits
Assay Genie offers a comprehensive range of apoptosis assays and ELISA kits to measure the key players in programmed cell death:
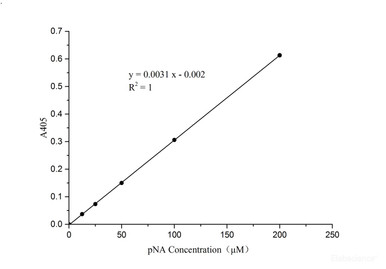
Caspase 3/7 Activity Assay Kit
Measure executioner caspase-3/7 activity, the convergence point of apoptosis.
View product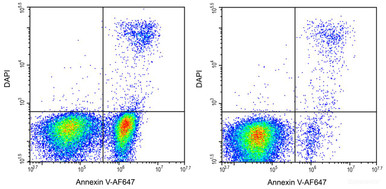
Annexin V-AF647 / DAPI Apoptosis Kit
Detect early apoptosis by Annexin V-AF647 and DAPI staining.
View product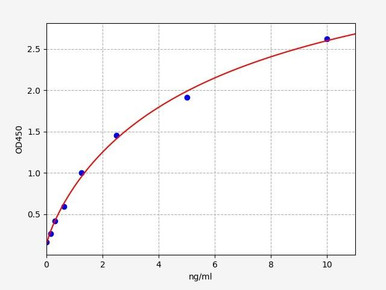
Human Caspase-8 ELISA Kit
Quantify caspase-8, the initiator of the extrinsic death-receptor pathway.
View product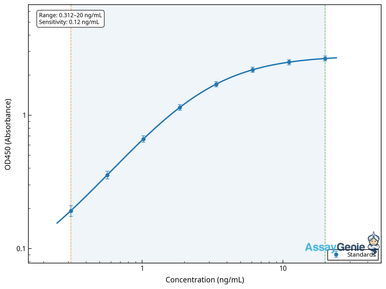
Human Caspase-9 ELISA Kit
Quantify caspase-9, the initiator of the intrinsic mitochondrial pathway.
View product
Intrinsic vs Extrinsic Apoptosis
The Extrinsic Apoptosis Pathway
The extrinsic pathway of apoptosis is initiated either by an extracellular signal that activates a death receptor (e.g., Fas, TNF), or by the release of Granzyme B and perforin from cytotoxic lymphocytes as part of the innate immune response to infection (Goping et al., 2003). Activation of the death receptors leads to their oligomerisation and recruitment of the cytosolic adaptor proteins (e.g,. FADD, TRADD), facilitating the assembly of the death-inducing signalling complex (DISC), which is comprised of the death receptors and associated adaptor proteins in conjunction with procaspase-8 (Ashkenazi et al., 1998).
Cell Death Receptors in Extrinsic Apoptosis
Caspase-8 Mediated Apoptosis
Upon activation caspase-8 can propagate the apoptotic signal by cleavage of further downstream effector caspases such as caspase-3 (Medema et al., 1997; Murphy et al., 2004). However, it has also been reported that caspase-8 can be activated in similar manner to caspase-9 where processing is not required and caspase-8 is activated by dimerisation (Boatright et al., 2003; Donepudi and Grutter, 2002).
Under some conditions caspases-8 cannot directly activate downstream caspases and hence there have been two cell types described both of which utilise the CD95 receptor /ligand complex (Scaffidi et al., 1998). In what have been termed type I cells, death is instigated by large amounts of active caspase-8 produced at the DISC, resulting in the direct cleavage of caspase-3 prior to loss of mitochondrial transmembrane potential. In comparison in the type II cells the activation of the death receptor pathway alone is insufficient to induce apoptosis. In these cells little DISC is formed and hence smaller amount of active caspase-8 is available and thus the apoptotic signal requires amplification by the simultaneous engagement of the mitochondrial pathway.
This pathway is carried out by caspase-8 mediated cleavage of a BH3-domain-containing Bcl-2 family member Bid. Active caspase-8 cleaves Bid to produce a truncated form (tBid), which either alone or in combination with other molecules induces mitochondria to release pro-apoptotic factors such as cytochrome c resulting in the formation of the apoptosome complex (Li et al., 1998; Luo et al., 1998).
The Intrinsic Apoptosis Pathway
What is MOMP?
MOMP stands for mitochondrial outer membrane permeabilisation. The process of apoptosis is mediated through the regulation of MOMP by pro-apoptotic and anti-apoptotic members of the Bcl-2 family of proteins (Green et al., 2004). MOMP is the point of no return where cells become irreversibly committed to cell death (Tait et al., 2010), and causes release of apoptogenic proteins such as cytochrome C, endoG, Smac/DIABLO, AIF-1, and Omi/HtrA2 from the intermembrane space into the cytoplasm.
Cytochrome C release into the cytoplasmic compartment leads to its interaction with Apaf-1, allowing the cleavage of procaspase-9, the formation of the Apaf-1/caspase-9 apoptosome complex, and the subsequent activation of effector caspases (Li et al., 1997; Zou et al., 1999). The fate of the cell is ultimately thought to be determined by the relative abundance and activity of the opposing Bcl-2 proteins.
Bcl-2 Family Proteins and Cell Death Regulation
Bax & Bak - MOMP Regulating Proteins
Bax and Bak function by forming homo- or hetero-dimeric pores in the outer mitochondrial membrane (MOMP) and catalysing the release of apoptogenic factors into the cytosol. Recent reports indicate that Bid, Bim, and PUMA are the primary activators of Bax/Bak dimerisation, with the other BH3-only proteins acting in a facilitatory fashion (Kim et al., 2006). Anti-apoptotic Bcl-2 members all possess BH domains 1-4, and all except Bcl-2A1 have a transmembrane region. Anti-apoptotic members act to prevent the formation of Bax/Bak dimers. The primary function of pro-apoptotic Bcl-2 proteins, therefore, is to form stable heterodimers with, and thus sequester, the anti-apoptotic members of the family, allowing Bax/Bak dimerisation and subsequent MOMP formation.
Two contrasting mechanisms for MOMP formation have been proposed. In the indirect model, all pro-apoptotic BH3-only proteins act only by interaction with anti-apoptotic members (Willis et al., 2005; Willis et al., 2007). In the direct model, BH3-only proteins are further subdivided into sensitisers (Bad, Bmf, Noxa) which antagonise the anti-apoptotic members, and activators (Bim, Puma, tBid) which directly promote Bak/Bax dimerisation (Galonek et al., 2006; Hyungjin et al., 2006).
BH3-only Proteins
Puma & Noxa Expression and Cell Death Activation
Puma and Noxa were identified through gene-expression profiling for targets of the tumour suppressor, p53, and from yeast two-hybrid screens using Bcl-2 as bait [Han et al., 2001; Nakano and Vousden, 2001; Oda et al., 2000; Yu et al., 2001]. The transcription factor, p53, transcriptionally upregulates Puma and Noxa following DNA damage, thus triggering apoptosis [Villunger et al., 2003a].
Puma and Noxa can bind and inhibit the anti-apoptotic Bcl-2 family members, Mcl-1, Bcl-2 and Bcl-XL, resulting in apoptosis [Nakano and Vousden, 2001; Oda et al., 2000; Yu et al., 2001]. Recently, Noxa has been implicated in H-Ras mediated autophagic cell death through increased expression and the displacement of Mcl- 1 from the autophagy regulator, Beclin-1 [Elgendy et al., 2010].
Bad Mediated Cell Death
The BH3-only protein, Bad, was originally identified as Bcl-XL interactor through yeast two-hybrid and -phage library screens, and was found to selectively dimerise with Bcl-2 and Bcl-XL, but not Mcl-1 [Yang et al., 1995]. In the presence of the survival factor, IL-3, Bad becomes phosphorylated by the Ser/Thr kinase, Akt, resulting in its binding to the 14-3-3 proteins, thus inhibiting its apoptotic function [Datta et al., 1997; Zha et al., 1996]. Phosphorylation of Bad by Cdk1 in post-mitotic neurons results in the dissociation of Bad from 14-3-3 proteins and subsequent cell death [Konishi et al., 2002].
Bim Phosphorylation and Anti-Apoptotic Effects
The BH3-only protein Bim has been extensively characterised as a mediator of intrinsic cell death pathways. Following cellular stresses such as UV irradiation, Taxol treatment and serum growth factor withdrawal, Bim localises to the mitochondria and inhibits the anti-apoptotic members of the Bcl-2 family, Bcl-2, Bcl-XL and Mcl-1, and activates Bak and Bax, resulting in MOMP and subsequent cell death [Kutuk and Letai, 2010].
ERK1/2 has been shown to phosphorylate BimEL at multiple sites [Weston et al., 2003]. Phosphorylation of BimEL by ERK1/2 following serum withdrawal results in its degradation by 20S proteasomes [Wiggins et al., 2011]. BimEL is phosphorylated by ERK1/2 at up to six different residues, three of which are within exon 3, an exon only expressed in BimEL. The phosphorylation of BimEL by ERK1/2 requires the DEF-domain (docking site for ERK, FXFP) within exon 3.
PD-1 and Apoptosis
Programmed cell death protein 1 (PD-1) can be found on the surface of T cells and plays an integral role in regulating immune responses by lessening T cell activation. By binding to either PD-L1 or PD-L2, it triggers apoptosis within the target T cells - a phenomenon known as programmed cell death.
PD-1-mediated apoptosis is a process by which the body's immune system can get rid of potentially hazardous activated T cells. When exposed to an antigen, T cells switch on their PD-1 expression and interact with either PD-L1 or PD-L2 present in Antigen Presenting Cells (APC) or other cell types found within the organism. This resultant interaction initiates programmed death of T cells thus preventing unwarranted systemic inflammation reactions from occurring.
Cancer cells employ the PD-1/PD-L1 pathway to escape detection by and hinder immune responses, allowing tumors to grow freely. By displaying high levels of PD-L1, malignant cells can restrain T cell activity and keep them from attacking the tumor. To counter this tactic, scientists invented immunotherapy drugs that target both proteins—and their results against a variety of cancers have been incredibly promising thus far!
Nevertheless, blocking PD-1/PD-L1 signaling can lead to unfavorable immune reactions such as autoimmune disorders and tissue damage. Therefore, it is essential to properly weigh the benefits and risks of applying PD-1/PD-L1 blockade when treating cancer patients.
To summarize, PD-1 is an integral part of regulating both T cell apoptosis and the immune system. The interactions between PD-1, PD-L1 and PD-L2 can have great ramifications for treating cancer as well as autoimmune conditions. Further research should be done to expand understanding on how exactly PD-1 induces apoptosis in order to create efficient therapies that target this pathway without causing any harm or side effects.
Caspase Apoptosis Pathway
Caspases can be classified into 3 different groups on the basis of their substrate specificity or length of their pro-domain (Grutter, 2000; Thornberry and Lazebnik, 1998). Group I caspases function primarily as mediators of inflammation and include caspases -1, -4, -5, -11 and -14. They are involved in the activation and processing of pro-inflammatory cytokines and appear not to have a main role in apoptosis. The Group I caspases prefer bulky hydrophobic residues in the P4 position, such as tyrosine or tryptophan. (Nicholson, 1999; Thornberry et al., 1997).
Caspase 3/7 Cell Death Signalling
Caspase 3 and 7 are two key apoptotic enzymes that work together to initiate cell death. Caspase 3 is responsible for cleaving proteins that lead to cell death, while caspase 7 activates caspase 3. Both of these enzymes are activated by pro-apoptotic signals, which can be triggered by a variety of stimuli, including DNA damage, oxidative stress, and apoptotic hormones. Once activated, caspase 3 and 7 work together to cleave a variety of cellular proteins, ultimately leading to cell death.
Caspase 3/7-mediated apoptosis is a critical process that helps to maintain tissue homeostasis and prevent the development of cancer. apoptosis is a type of programmed cell death that occurs in response to various stimuli, including DNA damage, oxidative stress, and apoptotic hormones. Caspase 3 and 7 are two key enzymes that work together to initiate apoptosis. Caspase 3 cleaves proteins that lead to cell death, while caspase 7 activates caspase 3.
Apoptosis is a critical process that helps to maintain tissue homeostasis and prevent the development of cancer. Caspase-mediated apoptosis is one type of programmed cell death that occurs in response to various stimuli, including DNA damage, oxidative stress, and apoptotic hormones.
Caspase 8 Activation, Cleavage Site & Pathway
Caspase 8 is an important regulator of apoptosis, and its activation is essential for the initiation of cell death. When apoptosis is triggered, caspase 8 cleaves and activates downstream proteins that lead to cell death. The caspase 8 cleavage site is located at amino acid Asp315, which is within the DEVD motif.
This cleavage site is essential for the activation of caspase 8 and subsequent apoptosis. Caspase 8 signaling leads to the activation of several downstream apoptotic proteins, including caspases 3, 6 and 7. These proteins then execute cell death by causing DNA fragmentation and cell shrinkage.
Caspase 9 Activation, Cleavage Site & Pathway
Caspase 9 is an important initiator caspase in the apoptotic pathway. It is activated by proteolytic cleavage at specific sites, which results in the release of its active catalytic domain. This then leads to the formation of an apoptosome, a key complex in the initiation of apoptosis.
Caspase 9 is activated by proteolytic cleavage at Asp-315 and Asp-330, which are located in the linker region between the large and small subunits. This results in the release of the active catalytic domain, which then goes on to form an apoptosome. The apoptosome is a key complex in the initiation of apoptosis, and is composed of caspase 9, caspase 3, and pro-apoptotic proteins such as cytochrome c.
When caspase 9 is cleaved at Asp-315, it results in the formation of an active tetramer that is capable of binding to and cleaving caspase 3. This results in the activation of caspase 3, which then leads to the proteolytic cleavage of a number of key proteins involved in cell death.
IAPS Regulation of Apoptosis
The IAPs regulate apoptosis by direct caspase inhibition (Deveraux and Reed, 1999; Deveraux et al., 1998; Riedl et al., 2001; Shiozaki et al., 2003). Interestingly, IAPs seem to be multifunctional and are not only involved in regulating apoptosis, but are also involved in receptor-mediated signalling, the cell cycle and ubiquitylation (Birkey Reffey et al., 2001; Hofer-Warbinek et al., 2000; Huang et al., 2000; Levkau et al., 2001; Lu et al., 2007; MacFarlane et al., 2002; Sanna et al., 2002; Sanna et al., 1998; Varfolomeev et al., 2007).
Marina Alberto, PhD, holds a robust academic background in Biotechnology, earning her Bachelor’s Degree and PhD in Science and Technology from Quilmes National University. Her research spans cancer immunotherapy, glycan profiling, and vaccine development, including innovative projects on pediatric leukemia diagnosis and cancer-associated carbohydrate-mimetic vaccines. She currently serves as a Technical Support and Sales Specialist at Assay Genie.
Frequently Asked Questions
What is apoptosis?
Apoptosis is programmed cell death — a tightly regulated process that removes damaged or unwanted cells without triggering inflammation.
What is the difference between the intrinsic and extrinsic pathways?
The extrinsic pathway is triggered by death-receptor ligands at the cell surface and activates caspase-8; the intrinsic (mitochondrial) pathway responds to internal stress via Bcl-2 family proteins and MOMP and activates caspase-9. Both converge on executioner caspases 3 and 7.
How is apoptosis different from necrosis?
Apoptosis is controlled and non-inflammatory, with orderly cell shrinkage and fragmentation, whereas necrosis is uncontrolled death that ruptures the membrane and causes inflammation.
How is apoptosis measured?
Common readouts include Annexin V staining, caspase-3/7, -8 and -9 activity assays, TUNEL, and ELISAs for regulators such as Bcl-2 and Bax.
Related reading
Studying cell death?
From caspase activity assays and Annexin V kits to Bcl-2, Bax and cytochrome c ELISAs, Assay Genie supplies the reagents behind apoptosis research — backed by expert technical support.
Browse ELISA Kits →Recent Posts
-
Red Blood Cell Peroxidase Interference in Competitive ELISA
QUICK ANSWER A researcher ran a competitive 2,3-bisphosphoglycerate (2,3-BPG) ELISA …4th Aug 2026 -
Sample Dilution Range Calculation for Competitive ELISA kits
Quick answer A competitive ELISA is running correctly when the blank (zero standard) gives the h …31st Jul 2026 -
Troubleshooting high ELISA concentrations
QUICK ANSWER If your ELISA returns sample concentrations that are orders of magnitud …30th Jul 2026